This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is Phylogeny?
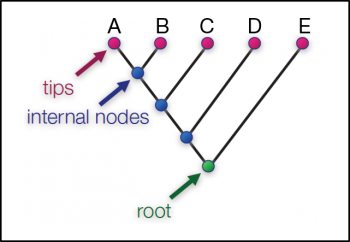
|
Phylogenetics is the study of evolutionary relationships among biological units. A phylogeny, or phylogenetic tree, illustrates these evolutionary relationships. Each branch on the tree shows the path of transmission of genetic information between generations, while each node represents a common ancestor to the branches it joins. The phylogeny is rooted at the common ancestor to all species in the phylogeny. [1]
|
Methods for Constructing a Phylogeny
|
Neighbor Joining
This is an algorithm that finds neighbors (sequence pairs) by minimizing total branch length at each level. This method is very efficient but is not the most accurate method. [2] |
Maximum Likelihood
The likelihood is the probability of the sequences occurring as they are given a model of their evolution on a particular tree. The more probable that the sequences would evolve given the tree, the more likely the tree is true. [3] |
Minimum Evolution
The minimum evolution model works to find the smallest total branch length for the entire phylogeny. This method works as long as the evolutionary distances are statistically unbiased. [4] |
Hepatic Lipase Phylogenetic Trees
Neighbor Joining Phylogeny
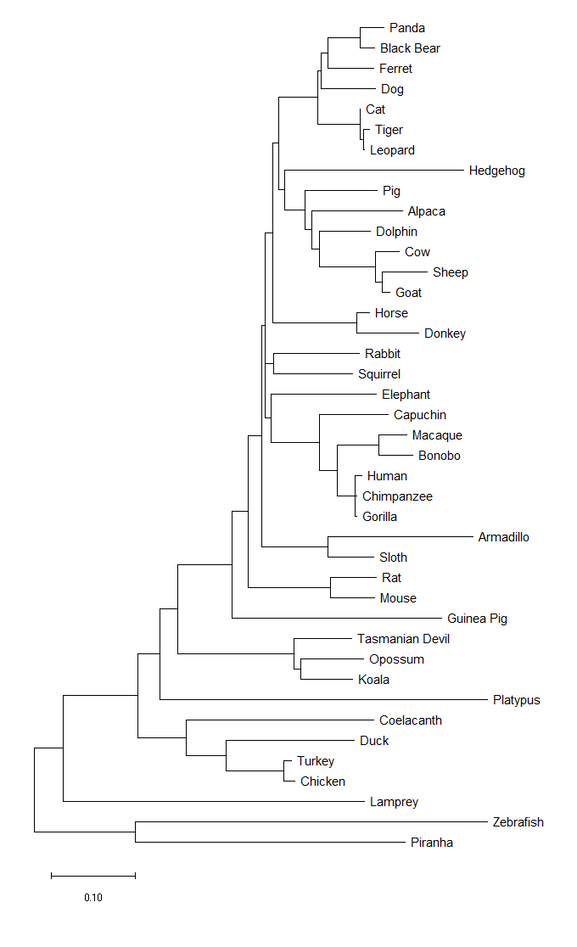
Figure 2. The evolutionary history was inferred using the Neighbor-Joining method [2]. The optimal tree with the sum of branch length = 5.46252372 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method [5] and are in the units of the number of amino acid substitutions per site. This analysis involved 41 amino acid sequences. All ambiguous positions were removed for each sequence pair (pairwise deletion option). There were a total of 644 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [6].
Maximum Likelihood Phylogeny
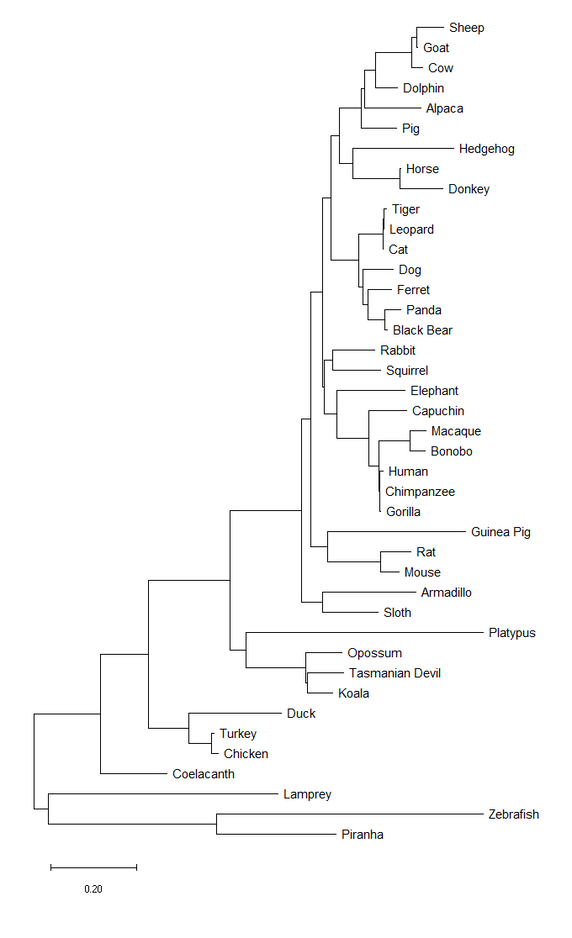
Figure 3. The evolutionary history was inferred by using the Maximum Likelihood method and JTT matrix-based model [7]. The tree with the highest log likelihood (-15781.88) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 41 amino acid sequences. There were a total of 644 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [6].
Minimum Evolution Phylogeny
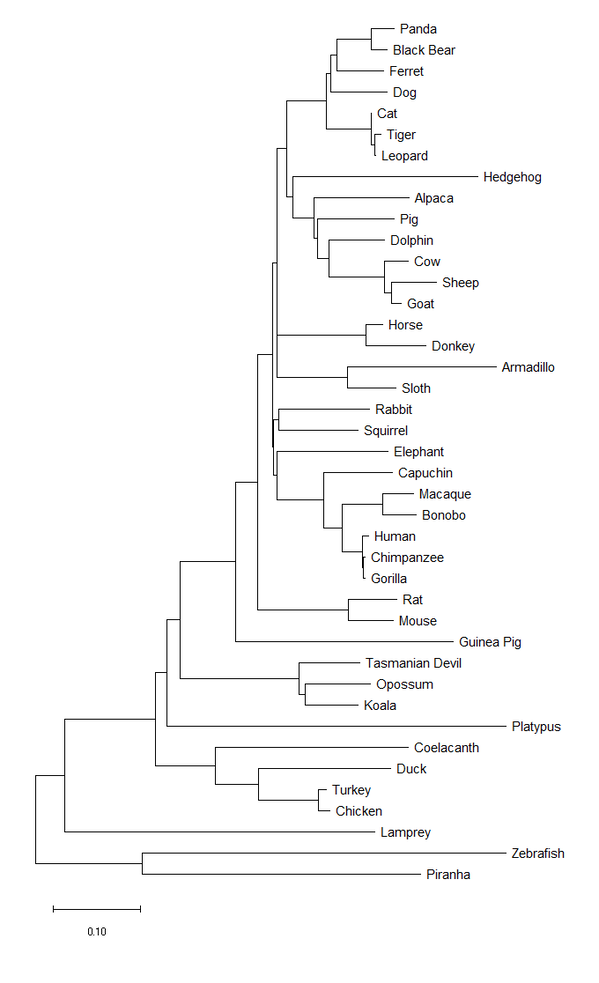
Figure 4. The evolutionary history was inferred using the Minimum Evolution method [8]. The optimal tree with the sum of branch length = 5.48301248 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method [9] and are in the units of the number of amino acid substitutions per site. The ME tree was searched using the Close-Neighbor-Interchange (CNI) algorithm [10] at a search level of 1. The Neighbor-joining algorithm [2] was used to generate the initial tree. This analysis involved 41 amino acid sequences. All ambiguous positions were removed for each sequence pair (pairwise deletion option). There were a total of 644 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [6].
Conclusions
Constructing a phylogeny can be very useful for determining relationships between proteins. This is useful for deciding which model organisms are similar enough to your protein of interest to be able to conduct valid experiments. For hepatic triaclglycerol lipase (HTGL), the method used to calculate the phylogenetic relationships not only affected the predicted evolutionary distances but also the predicted relationships.
References
[1] Aspects of Phylogeny. (2016, June 08). Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogeny/aspects-phylogenies
[2] Saitou N. and Nei M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4:406-425.
[3] Maximum Likelihood. (n.d.). Retrieved from https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
[4] Theoretical foundation of the minimum-evolution method of phylogenetic inference. (1993). Molecular Biology and Evolution. doi:10.1093/oxfordjournals.molbev.a040056
[5] Zuckerkandl E. and Pauling L. (1965). Evolutionary divergence and convergence in proteins. Edited in Evolving Genes and Proteins by V. Bryson and H.J. Vogel, pp. 97-166. Academic Press, New York.
[6] Kumar S., Stecher G., Li M., Knyaz C., and Tamura K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution 35:1547-1549.
[7] Jones D.T., Taylor W.R., and Thornton J.M. (1992). The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences 8: 275-282.
[8] Rzhetsky A. and Nei M. (1992). A simple method for estimating and testing minimum evolution trees. Molecular Biology and Evolution 9:945-967.[9] Zuckerkandl E. and Pauling L. (1965). Evolutionary divergence and convergence in proteins. Edited in Evolving Genes and Proteins by V. Bryson and H.J. Vogel, pp. 97-166. Academic Press, New York.
[10] Nei M. and Kumar S. (2000). Molecular Evolution and Phylogenetics. Oxford University Press, New York.
Non-linked Figures:
Header
[2] Saitou N. and Nei M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4:406-425.
[3] Maximum Likelihood. (n.d.). Retrieved from https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
[4] Theoretical foundation of the minimum-evolution method of phylogenetic inference. (1993). Molecular Biology and Evolution. doi:10.1093/oxfordjournals.molbev.a040056
[5] Zuckerkandl E. and Pauling L. (1965). Evolutionary divergence and convergence in proteins. Edited in Evolving Genes and Proteins by V. Bryson and H.J. Vogel, pp. 97-166. Academic Press, New York.
[6] Kumar S., Stecher G., Li M., Knyaz C., and Tamura K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution 35:1547-1549.
[7] Jones D.T., Taylor W.R., and Thornton J.M. (1992). The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences 8: 275-282.
[8] Rzhetsky A. and Nei M. (1992). A simple method for estimating and testing minimum evolution trees. Molecular Biology and Evolution 9:945-967.[9] Zuckerkandl E. and Pauling L. (1965). Evolutionary divergence and convergence in proteins. Edited in Evolving Genes and Proteins by V. Bryson and H.J. Vogel, pp. 97-166. Academic Press, New York.
[10] Nei M. and Kumar S. (2000). Molecular Evolution and Phylogenetics. Oxford University Press, New York.
Non-linked Figures:
Header
